

Sleep is the output
A clinical framework for addressing upstream drivers of fragmented sleep

What started as a fun article to help a couple of friends compete for the winning sleep score has turned into an unexpected series on the science of sleep. We began with Why your sleep score sucks, where I explored what our wearables can (and cannot) tell us about our nightly recovery.
A reader’s question then led to Restoring sleep architecture, where I unpacked hypnograms, sleep architecture, and the fascinating dance between deep and REM sleep throughout the night.
This next article gets a bit more clinical because a clinical question was asked by a reader. So this is intended to help practitioners think through common causes of disrupted sleep architecture. There’s something here, though, for everyone.
I’ve created a patient guide that summarizes the primary drivers of sleep architecture disruption discussed in this article. It is designed to facilitate patient-provider conversations and may be shared directly with patients or adapted for use within clinical practice.
The following is intended for educational purposes only, and although it is evidence-supported, it is not medical advice and does not replace the advice of your medical provider. If you are suffering from sleep deprivation, insomnia, prolonged stress, or other symptoms discussed in this article, please seek the advice of a medical professional.
Our sleep is a precisely orchestrated biological program that cycles through distinct stages every night: light sleep (N1/N2), slow-wave deep sleep (N3), and rapid eye movement (REM) sleep. As discussed in Restorative sleep stages, each stage serves unique physiological functions that cannot be fully replicated by the others.
These stages are measured directly using polysomnography (PSG), the gold standard for sleep assessment, or estimated by consumer wearables such as the RingConn, Apple Watch, Oura Ring, and Whoop. Data collection is displayed in a hypnogram, a graph illustrating the rhythmic progression through sleep stages across the night. Together, these recurring cycles comprise sleep architecture.
In healthy adults, sleep architecture follows a relatively predictable pattern. After falling asleep, individuals typically progress from N1 sleep into N2 sleep and then into N3 slow-wave (deep) sleep before cycling back through lighter stages and entering REM sleep. This sequence repeats throughout the night, with N2 sleep comprising the largest proportion of total sleep time, while N3 and REM sleep occupy smaller but physiologically important portions of the sleep period.
Although the exact distribution varies by age and individual factors, healthy sleep is generally marked by a stable cycling pattern between NREM and REM stages rather than frequent fragmentation or prolonged wakefulness [14].
When that architecture becomes disrupted, the consequences extend far beyond feeling tired the next day. Alterations in sleep-stage distribution have been associated with impaired cognitive performance, metabolic dysfunction, cardiovascular risk, immune dysregulation, and reduced recovery capacity.
A landmark 2024 prospective cohort study published in Sleep helped reframe this discussion. demonstrating that sleep regularity, the day-to-day consistency of sleep-wake timing, is a stronger predictor of mortality risk than sleep duration [18].
A subsequent 2025 review in Circulation Research confirmed that greater sleep irregularity is consistently associated with worse cardiometabolic outcomes across diverse populations [5].
The mechanism that disrupts sleep architecture over time is the same one that disrupts regularity, and this irregularity carries a measurable mortality risk independent of how many hours the patient reports sleeping.
In many common clinical presentations, sleep architecture disruption is dominated by one or more of three upstream physiological patterns: hypothalamic-pituitary-adrenal (HPA) axis dysregulation, circadian misalignment, and metabolic dysfunction. These patterns frequently coexist and interact with other sleep-disrupting conditions.
Driver One: HPA Axis Dysregulation
Mechanism
The stress response and restorative sleep are physiologically antagonistic. Sleep onset requires a precipitous drop in cortisol and central norepinephrine (NE), both of which must remain suppressed through the first two-thirds of the night for slow-wave sleep (SWS) to occur and be maintained. When the HPA axis is chronically activated, this suppression fails.
The pathway begins in the paraventricular nucleus (PVN) of the hypothalamus, which releases corticotropin-releasing hormone (CRH) in response to psychological, physiological, or inflammatory stressors. CRH stimulates pituitary adrenocorticotropic hormone (ACTH) secretion, which in turn drives adrenal cortisol release. Elevated nocturnal cortisol acts directly on sleep circuitry: it suppresses the growth hormone (GH) pulse that normally anchors N3 (our deep sleep) in the first sleep cycle, blunts slow-wave activity (SWA) amplitude, and fragments the continuity required to sustain deep non-rapid eye movement (NREM) sleep.
Nightlong recordings confirm that awakenings are positively correlated with ACTH levels, and both ACTH and cortisol are inversely related to slow-wave sleep. This pattern is consistent with CRH hypersecretion [7].
The locus coeruleus (LC) is the second critical node.
A 2025 landmark study published in Nature Neuroscience provided the first mechanistic proof of what clinicians have observed empirically for decades. Using optogenetics and fiber photometry, the authors demonstrated that infra-slow fluctuations in LC neuronal activity directly partition NREM sleep into two distinct brain-autonomic states that govern the entire NREM-REM cycle [12].
High LC activity, precisely the state produced by stress, induces subcortical-autonomic arousal that facilitates cortical microarousals and blocks REM entries entirely. Low LC activity is an obligatory prerequisite for NREM-to-REM transitions. Critically, a stress-promoting wakefulness experience was shown to elevate LC activity during subsequent NREM sleep, producing more microarousal-induced NREM fragmentation and delayed REM onset.
This is not a correlation. It is the identified circuit mechanism by which stress suppresses REM [12].
The result is a sleep architecture characterized by excess N1/N2, reduced SWS, fragmented or absent REM, and a hyperarousal phenotype that persists even when sleep duration appears adequate on actigraphy or upon self-report.
Psychosocial stress and its cognitive correlates, rumination (perseverative past-focused cognition) and worry (anticipatory threat processing), produce the same neuroendocrine signature with documented associations in [8]:
Prolonged sleep onset latency
Shorter and more fragmented sleep
Excess N1
Reduced REM
Reduced SWS
Prolonged SWS latency
This means a patient does not require an objectively measurable stressor, as the internal stress response is sufficient to produce full architectural disruption.
Clinical Fingerprint
Sleep initiation is often preserved or mildly impaired
Early morning awakening, characteristically between 2:00–4:00 AM, with cognitive hyperarousal and “the mind is already running”
Sleep is described as light, thin, or unrefreshing despite adequate duration
Night-to-night variability that correlates with identifiable stressors
Suppressed nocturnal heart rate variability (HRV); elevated resting heart rate during sleep (wearable data is clinically useful here)
Blunted cortisol awakening response (CAR) in chronic HPA exhaustion states; the patient wakes groggy, not alert, and takes 60+ minutes to feel functional
First-line treatment
Cognitive Behavioral Therapy for Insomnia (CBT-I) is the evidence-based standard and can be offered before or alongside any pharmacologic intervention. Meta-analyses and five clinical guidelines now unanimously recommend multicomponent CBT-I as first-line treatment for chronic insomnia [19].
The sleep restriction and stimulus control components directly address hyperarousal and conditioned arousal, respectively, while cognitive restructuring targets the perseverative cognition that sustains HPA activation. A 2025 FDA-authorized digital CBT-I trial (SleepioRx) confirmed sustained efficacy at 6 months in a decentralized nationwide randomized controlled trial [6].
When to refer
Refer to psychiatry or psychology if CBT-I is unavailable or has failed after 6–8 weeks; if comorbid anxiety disorder, post-traumatic stress disorder (PTSD), or major depressive disorder (MDD) is suspected; where clinically indicated, assessment of diurnal cortisol rhythm may provide additional information regarding neuroendocrine regulation, although interpretation remains context dependent.
Driver Two: Circadian Misalignment
Mechanism
The circadian system gates specific sleep stages to specific biological times of night through a precisely orchestrated program governed by the suprachiasmatic nucleus (SCN) of the hypothalamus. The SCN receives photic input from intrinsically photosensitive retinal ganglion cells (ipRGCs) via the retinohypothalamic tract and synchronizes peripheral clocks in virtually every tissue, including the brainstem circuits that govern sleep-stage timing [10].
REM sleep is the most clock-dependent stage.
Within Borbély’s two-process model of sleep regulation, process S (homeostatic sleep pressure, driven by adenosine accumulation) governs SWS, which is why SWS is more resilient to timing shifts.
Process C (the circadian pacemaker), operating through the SCN, tightly gates REM propensity to the final third of the biological sleep period [2].
This is not a passive effect: the SCN actively promotes REM during the subjective late-night/early-morning window and suppresses it at other times. A patient with a biological sleep midpoint of 4:00 AM who is forced awake at 6:00 AM by an alarm or other means is losing two or more hours of peak REM propensity every single night because process C has not yet permitted maximal REM expression.
The architecture consequence is REM compression or frank REM deprivation, with SWS relatively preserved.
REM sleep is typically the earliest and most sensitive casualty of circadian misalignment because REM expression is tightly gated by the circadian clock, whereas slow-wave sleep remains more strongly governed by accumulated sleep pressure. Although SWS is generally more resilient, chronic or severe circadian misalignment can eventually fragment overall sleep architecture and reduce slow-wave sleep quality [27].
The chronic discordance between a person’s endogenous chronotype and their socially imposed schedule (social jetlag) is now recognized as a major population-level source of circadian misalignment. A 2023 population-representative longitudinal study in Sleep found that social jetlag is independently associated with adverse cardiovascular and metabolic biomarkers, including unfavorable lipid profiles, after controlling for chronotype, age, sex, and body mass index [16].
Shift workers carry a 40% increased risk of type 2 diabetes compared with non-shift workers, driven in part by circadian-mediated glucose dysregulation and sleep architecture fragmentation [14].
The 2025 Circulation Research review established that internal deviations in sleep timing, not just sleep duration, represent the most prevalent cause of circadian disruption at the population level, and that sleep irregularity may be a more significant predictor of cardiometabolic disease risk than sleep duration itself [5].
A patient who “sleeps enough hours” may be incurring significant metabolic and mortality risks due to timing irregularity alone.
Emerging evidence also demonstrates that combining CBT-I with circadian rhythm support (consistent scheduling, timed light, meal timing) yields superior outcomes compared with either approach alone [13].
Clinical Fingerprint
Sleep quality and architecture improve substantially on free days when no alarm intervenes (the “vacation test”)
Disproportionate sleep inertia on required-schedule mornings causes dense, prolonged grogginess, suggesting waking during biological night
Natural sleep midpoint (estimated as the midpoint between spontaneous sleep onset and natural waking) is significantly later than required wake time demands
Patient identifies as a “night owl” and performs better cognitively and physically in evening hours
Daytime sleepiness despite adequate reported duration (Epworth Sleepiness Scale [ESS] ≥10 warrants formal evaluation)
Mood and energy are markedly better when sleeping on a natural schedule
First-line treatment
Chronotherapy targets realignment of internal circadian rhythms with the external light-dark cycle and the patient’s required schedule. Four levers are available: light, melatonin timing, meal timing, and schedule consistency.
Timed morning bright light: The American Academy of Sleep Medicine (AASM) Clinical Practice Guideline for delayed sleep-wake phase disorder (DSWPD) identifies timed bright light exposure as a primary treatment. Morning light (10,000 lux, 30–60 minutes) delivered at biological dawn begins to advance the phase response curve (PRC). Light exposure at the wrong time will shift the clock in the wrong direction; timing relative to the individual’s dim light melatonin onset (DLMO) is key [1].
Timed melatonin: The AASM guideline recommends timed exogenous melatonin for DSWPD. Melatonin administered 5–6 hours before the patient’s current sleep onset (i.e., in the late afternoon for a severely delayed patient) produces the maximum phase-advance on the PRC [1]. The standard dose is 0.3–0.5 mg; higher doses do not produce a proportionally greater phase shift and may cause next-day grogginess.
Schedule anchoring: Consistency of wake time, even on weekends, is the single most powerful behavioral anchor for circadian entrainment. A 2025 preprint narrative review confirmed that combining CBT-I with circadian rhythm support yields superior outcomes compared with either alone [13].
Meal timing: Time-restricted eating (TRE) aligned with the light phase (front-loading calories to morning/midday) supports peripheral circadian entrainment and reduces social jetlag effects on metabolic health.
Peripheral to the master clock are tissues that have their own rhythmic clocks. These include the liver, muscle, adipose, pancreas, and the gut (among many others) and run a self-sustaining transcription-translation feedback loop driven by CLOCK/BMAL1 protein cycling.
These peripheral clocks cannot directly sense light so they entrain primarily to feeding-fasting cycles, making the timing of food intake one of the most potent zeitgebers (”time-givers”) available outside of light itself.
When meals are shifted late, as is typical for individuals with social jetlag or night owls, peripheral clocks in the liver and gut decouple from the central SCN clock. This internal desynchrony between brain time and organ time is metabolically costly because it produces insulin resistance, blunts postprandial glucose clearance, elevates triglycerides, and disrupts the nocturnal suppression of hepatic glucose output [29], which as been demonstrated in controlled feeding studies in humans.
TRE consolidates the feeding-fasting signal and strengthens peripheral clock entrainment independent of caloric restriction. The circadian relevance is distinct from the metabolic effects of calorie reduction, which has made TRE an increasingly studied tool for circadian medicine specifically.
When to refer
Refer to a sleep medicine specialist if DSWPD, advanced sleep-wake phase disorder (ASWPD), non-24-hour sleep-wake disorder, or shift work disorder is suspected and fails to respond to behavioral chronotherapy within 4–6 weeks. Actigraphy over 14 days can be an appropriate objective assessment tool prior to referral. Consider salivary DLMO testing to personalize melatonin timing; home-based DLMO kits are increasingly available and clinically actionable.
Driver Three: Metabolic Dysfunction
Mechanism
This is the most underappreciated and underdiagnosed driver of sleep architecture disruption in primary care and the one with the highest downstream systemic stakes. The relationship is bidirectional and self-amplifying: metabolic dysfunction degrades sleep architecture, which worsens insulin sensitivity, which further disrupts sleep.
A January 2025 review of current evidence confirmed that insufficient sleep and poor sleep quality are independently associated with increased risks of obesity, type 2 diabetes, and cardiovascular disease, with bidirectionality well established in prospective cohort data [21].
The specific sleep-stage mechanism is now well characterized.
In a 2023 study published in Cell Reports Medicine, Vallat et al. examined over 600 people with overnight polysomnography and next-morning glucose and insulin measurements, then replicated their findings in an independent cohort of over 1,900 adults.
The key finding was that the coupling of NREM sleep spindles and slow oscillations during N3 (the electrophysiological signature of deep sleep) was the strongest predictor of next-day peripheral glucose control, outperforming traditional sleep metrics, including total sleep time and sleep efficiency [17].
The mechanism operates through altered insulin sensitivity rather than pancreatic beta cell function, running from the sleeping brain through cardiac autonomic pathways to peripheral glucose regulation [17].
A 2024 review in the Journal of Clinical Medicine further confirmed that diabetes is consistently associated with reduced SWS even in the absence of sleep-disordered breathing, and that selective SWS suppression, without reducing total sleep time, leads to significant increases in insulin resistance and decreased glucose tolerance [11].
Glucose instability and nocturnal arousal: Blood glucose dips during the early morning hours trigger a sympathoadrenal counterregulatory cascade of epinephrine and cortisol release, producing an arousal pattern clinically indistinguishable from HPA-driven insomnia.
A patient may wake at 3:00–4:00 AM, feeling physiologically alert but not cognitively anxious from a glucose recovery response. Research using continuous glucose monitoring (CGM) during polysomnography has demonstrated that patients with moderate-to-severe obstructive sleep apnea (OSA) exhibit rising blood glucose levels after sleep onset, directly linking nocturnal glucose dysregulation to sleep-architecture disruption [20].
Sleep-disordered breathing: OSA and upper airway resistance syndrome (UARS) fragment both SWS and REM through repeated micro-arousals that typically last 3–15 seconds. The patient may never consciously register it, but that prevents sustained stage consolidation. The sleep architecture of OSA is characterized by increased N1 and N2 sleep and decreased SWS and REM sleep, with arousal frequency as the primary driver [15].
Many patients do technically enter N3 and REM sleep, but repeated respiratory events and microarousals prevent these stages from being sustained long enough to deliver their normal restorative benefits [28].
Intermittent hypoxia and frequent arousal increase sympathetic tone, oxidative stress, systemic inflammation, and hormonal imbalances, producing insulin resistance and beta cell dysfunction through pathways distinct from chronic sleep deprivation.
Systemic inflammation: IL-6 and TNF-α are elevated in disorders associated with excessive daytime sleepiness, including [7]:
Sleep apnea
Narcolepsy
Idiopathic hypersomnia
Obesity-mediated chronic low-grade inflammation shifts sleep toward lighter stages and blunts slow-wave amplitude through these cytokine pathways.
Clinical Fingerprint
Sleep consistently feels unrefreshing regardless of duration; the patient sleeps 8 hours and wakes exhausted
Fatigue that tracks with meals, carbohydrate load, or weight fluctuation
Nocturnal arousal at 3:00–4:00 AM that feels physiological rather than cognitive; the patient wakes alert but not anxious
Morning headaches, dry mouth, or sore throat on waking
Snoring reported by bed partner, even intermittently
ESS ≥10 despite reported adequate sleep duration
BMI >30, neck circumference >17 inches (male) or >16 inches (female), or retrognathia on exam
Resistant hypertension or atrial fibrillation; OSA association is strong enough to warrant screening automatically
First-line treatment
Metabolic stabilization:
Stabilize nocturnal glucose: Reduce refined carbohydrate load, particularly in the evening. Consider CGM for 10-14 days to assess whether nocturnal awakening coincides with overnight glucose fluctuations. In patients with suspected nocturnal glucose instability, adjustment of evening meal composition may be considered, although evidence remains limited, and individualized assessment is warranted.
HOMA-IR: Assess insulin resistance as a baseline metabolic marker, particularly in patients with unrefreshing sleep and fatigue-meal correlation.
Inflammatory biomarkers, including hs-CRP and, where available, IL-6, may provide additional context regarding systemic inflammatory burden.
Sleep-disordered breathing:
Home sleep testing (HST) is an appropriate first-line for suspected uncomplicated OSA in adults without significant cardiopulmonary comorbidity. In-laboratory polysomnography (PSG) is indicated when HST is negative but clinical suspicion remains high, when UARS is suspected, when significant cardiac or pulmonary comorbidity is present, or when full staging data are needed [9].
Continuous positive airway pressure (CPAP) initiation for confirmed OSA produces a well-documented rebound in both SWS and REM, with REM percentage exceeding normal values in the first weeks of treatment as architecture reconsolidates [15]. This rebound is expected and should be communicated to the patient.
UARS without classic apneic events, normal oxygen saturation, profoundly unrefreshing sleep, is routinely missed on HST and requires full PSG for diagnosis. Consider it in patients with the metabolic fingerprint above and a negative home study.
When to refer
Refer to sleep medicine for PSG when HST is negative, and suspicion remains, when UARS is suspected, when ESS ≥10 fails to respond to initial interventions, when REM behavior disorder (RBD) is suspected, or when the clinical picture remains unexplained after thorough primary evaluation. Refer to endocrinology if significant insulin resistance, cortisol dysregulation, or thyroid dysfunction is identified on workup.
The Interaction Problem
These three drivers do not operate in isolation. Chronic stress promotes insulin resistance through cortisol-mediated hepatic glucose output and peripheral insulin receptor downregulation. Metabolic dysfunction worsens circadian timing by disrupting the hormonal milieu on which peripheral clocks depend. Circadian disruption elevates cortisol through SCN-HPA crosstalk, closing the loop. The cascade can initiate from any node and can propagate.
Common Mimics and Amplifiers
HPA-axis dysregulation, circadian misalignment, and metabolic dysfunction account for many presentations of disrupted sleep architecture, but several additional conditions can independently fragment sleep or amplify disruption arising from these primary drivers. A brief orientation follows; thorough treatment of each is beyond the scope of this article.
Restless Legs Syndrome and Periodic Limb Movement Disorder
These are two important differential considerations in patients with sleep-maintenance insomnia, non-restorative sleep, or unexplained daytime sleepiness. Repetitive limb movements during sleep produce recurrent micro-arousals that impair continuity even when total sleep duration appears adequate [22].
The 2025 AASM clinical practice guideline represents a significant shift: routine assessment of iron status is now emphasized, dopamine agonists are deprioritized due to augmentation risk, and iron replacement (when indicated) alongside alpha-2-delta ligands are now the favored approaches. Low ferritin, an evening urge to move the legs, and a bed partner reporting repetitive movements are the key clinical signals.
Menopause and Perimenopause
These mid-life chapters produce a distinct sleep disruption phenotype characterized primarily by increased nighttime awakenings and greater wake after sleep onset (WASO), with reduced sleep efficiency — and notably, these disturbances may occur even in the absence of vasomotor symptoms [23].
Emerging literature further links menopausal sleep disruption to increased prevalence of sleep-disordered breathing, creating overlap with the metabolic and circadian mechanisms above [24]. New-onset insomnia during midlife in women warrants this differential.
Medication and Substance-Induced Disruption
SSRIs and SNRIs reliably prolong REM latency and alter REM expression in susceptible patients. Systemic corticosteroids commonly increase CNS activation, prolonging sleep onset and increasing nighttime awakenings. Stimulants, nicotine, and sympathomimetics delay sleep onset and increase nocturnal arousal through noradrenergic signaling.
Alcohol deserves particular attention because it reliably creates the illusion of improved sleep. While it often reduces sleep-onset latency, controlled studies and systematic reviews consistently show that alcohol disrupts architecture during the second half of the night: it delays REM expression, reduces REM duration, increases fragmentation, and exacerbates sleep-disordered breathing in susceptible individuals [25, 26].
A patient who reports “sleeping fine” with a nightly drink and wakes unrefreshed is a candidate for a structured trial without it.
Restoring Sleep Architecture for Patients
Sleep medications and supplements may have a role in selected patients, but they do not address the underlying reason sleep architecture became disrupted in the first place. Effective intervention begins with pattern recognition: identifying whether disruption is being driven by circadian misalignment, glycemic instability, stress physiology, sleep-disordered breathing, medication effects, or another upstream cause.
The goal is not simply to increase deep sleep or REM sleep. The goal is to restore the conditions that allow healthy sleep architecture to emerge naturally.
To support that process, I have created a patient-facing guide that organizes common causes of sleep architecture disruption into the three-driver framework described in this article. Clinicians may share it directly or adapt the recommendations to fit their own practice and treatment philosophy.
You can also download it here.
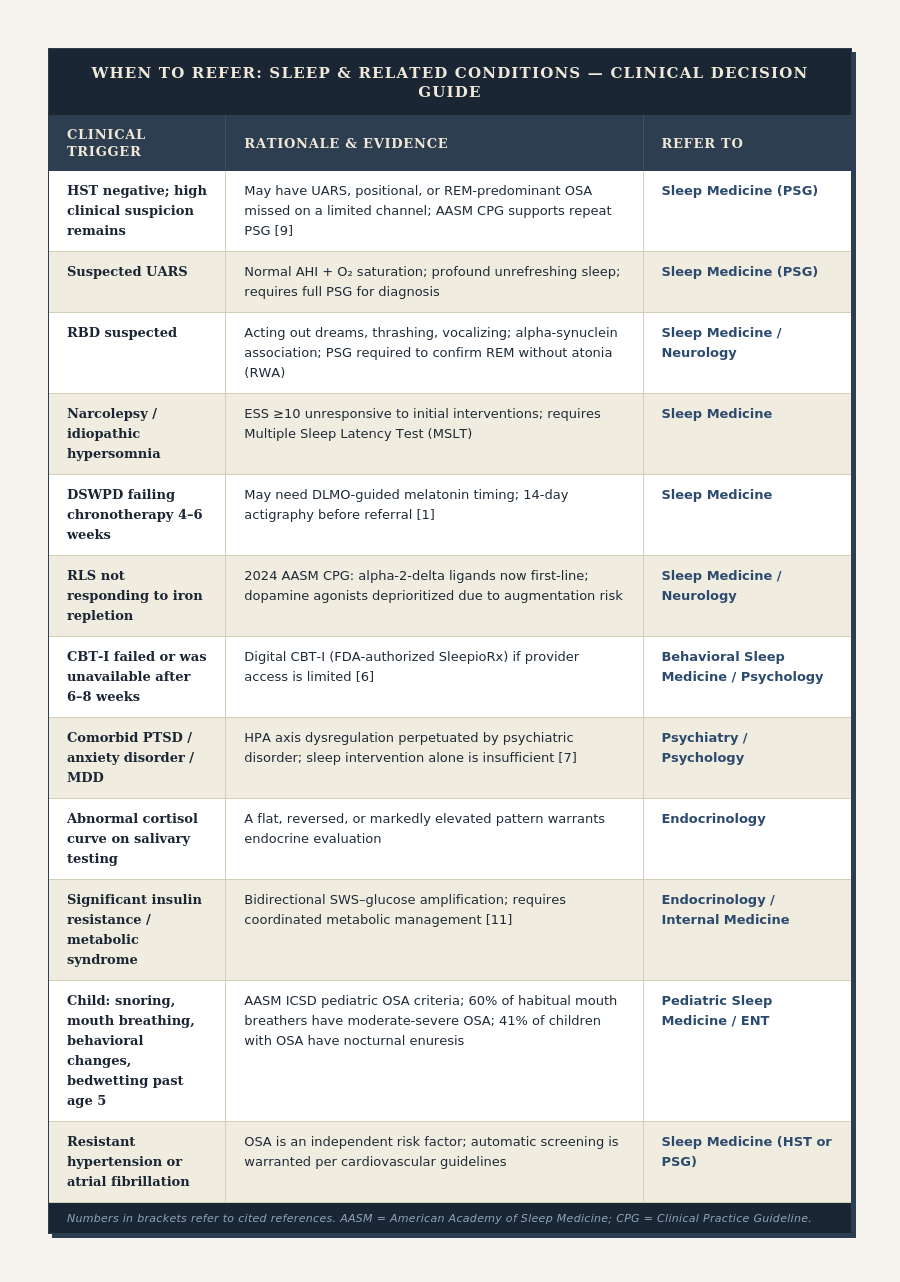
Clinical Patient Guide
To support clinical pattern recognition, I built an accompanying patient guide, which organizes common causes of sleep architecture disruption into three broad categories: stress physiology, circadian misalignment, and metabolic dysfunction. While many patients will exhibit features of more than one category, these patterns can provide a useful starting point for clinical investigation and patient education.
What’s Next?
Over the past several articles, we’ve taken a progressively deeper look at sleep. We began by exploring what healthy sleep patterns actually look like and how sleep architecture changes throughout the night. We then examined hypnograms and wearable data, learning both the value and limitations of the sleep metrics many of us check each morning. Finally, we turned our attention to restorative sleep itself, focusing less on scores and stages and more on the biological recovery processes that determine how we feel when we wake up.
Through this happy accident of a sleep series, I have been waiting to have an important conversation with you.
Sleep is not the system in charge, it is an output. It is not even the beginning of the story.
So we must now move beyond sleep and into the fascinating world of circadian biology, chronotypes, and the molecular clocks ticking inside nearly every cell of the human body.
──────────────────────────────
If this was useful, here’s where to go next:
→ You’re navigating chronic illness and want a clear roadmap: go here
→ You lead a clinic and want to bring this education to your patients: grab the sample curriculum here
→ You run a retreat and want to add science-backed depth to your program: go here
──────────────────────────────
References
1. Auger, R. R., Burgess, H. J., Emens, J. S., Deriy, L. V., Thomas, S. M., & Sharkey, K. M. (2015). Clinical practice guideline for the treatment of intrinsic circadian rhythm sleep-wake disorders: Advanced sleep-wake phase disorder (ASWPD), delayed sleep-wake phase disorder (DSWPD), non-24-hour sleep-wake rhythm disorder (N24SWD), and irregular sleep-wake rhythm disorder (ISWRD). Journal of Clinical Sleep Medicine, 11(10), 1199–1236. https://doi.org/10.5664/jcsm.5100
2. Bes, F. (2025). Treatise on an alternative perspective on the two-process model of sleep regulation. npj Biological Timing and Sleep, 2, Article 38. https://doi.org/10.1038/s44323-025-00038-0
3. Bonilla, D. A., Moreno, Y., Gho, C., Petro, J. L., Odriozola-Martínez, A., & Kreider, R. B. (2026). Effects of multi-herb and ashwagandha root formulas on stress modulation: A randomized, double-blind, placebo-controlled clinical study. Trials, 27, Article 495. https://doi.org/10.1186/s13063-026-09495-9
4. Brendler, T. (2025). Evaluation of potential hormonal activities of Ashwagandha (Withania somnifera). Phytotherapy Research. Advance online publication. https://doi.org/10.1002/ptr.70155
5. Chung, J., Goodman, M. O., Huang, T., Castro-Diehl, C., Redline, S., & Bhatt, D. L. (2025). Sleep irregularity, circadian disruption, and cardiometabolic disease risk. Circulation Research. Advance online publication. https://doi.org/10.1161/CIRCRESAHA.125.325613
6. Espie, C. A., Emsley, R., Kyle, S. D., Gordon, C., Drake, C. L., Siriwardena, A. N., Cape, J., Ong, J. C., Carr, A., Wills, G., & Luik, A. I. (2025). The effectiveness of digital cognitive behavioral therapy to treat insomnia disorder in US adults: Nationwide decentralized randomized controlled trial. JMIR Mental Health, 12, e84323. https://doi.org/10.2196/84323
7. Irwin, M. R., Piber, D., & Olmstead, R. (2025). Sleep and psychiatric disorders: Bidirectional interactions and shared mechanisms. PLOS Mental Health, 2(12), e0000531. https://doi.org/10.1371/journal.pmen.0000531
8. Kalmbach, D. A., Cuamatzi-Castelan, A. S., Tonnu, C. V., Tran, K. M., Anderson, J. R., Roth, T., & Drake, C. L. (2018). Hyperarousal and sleep reactivity in insomnia: Current insights. Nature and Science of Sleep, 10, 193–201. https://doi.org/10.2147/NSS.S138823
9. Kapur, V. K., Auckley, D. H., Chowdhuri, S., Kuhlmann, D. C., Mehra, R., Ramar, K., & Harrod, C. G. (2017). Clinical practice guideline for diagnostic testing for adult obstructive sleep apnea: An American Academy of Sleep Medicine clinical practice guideline. Journal of Clinical Sleep Medicine, 13(3), 479–504. https://doi.org/10.5664/jcsm.6506
10. Kolbe, I., Linde, K., Oster, H., & Keshet, G. (2024). Circadian dysfunction and cardio-metabolic disorders in humans. Frontiers in Endocrinology, 15, Article 1328139. https://doi.org/10.3389/fendo.2024.1328139
11. Mao, Y. (2024). Sleep architecture changes in diabetes. Journal of Clinical Medicine, 13(22), Article 6851. https://doi.org/10.3390/jcm13226851
12. Osorio-Forero, A., Foustoukos, G., Cardis, R., Cherrad, N., Devenoges, C., Fernandez, L. M. J., & Lüthi, A. (2025). Infraslow noradrenergic locus coeruleus activity fluctuations are gatekeepers of the NREM–REM sleep cycle. Nature Neuroscience, 28, 84–96. https://doi.org/10.1038/s41593-024-01822-0
13. Preprints.org. (2025, November 28). Circadian-based sleep interventions in clinical applications: A narrative review (Preprint). https://doi.org/10.20944/preprints202511.2169.v1
14. Rogers, M. A. (2024). The effects of sleep disruption on metabolism, hunger, and satiety, and the influence of psychosocial stress and exercise: A narrative review. Diabetes/Metabolism Research and Reviews, Article e3667. https://doi.org/10.1002/dmrr.3667
15. Shen, C. H., Huang, Y. S., Tseng, W. J., Yu, C. C., Lin, Y. N., Wang, C. Y., & Guilleminault, C. (2021). Rapid eye movement sleep and slow wave sleep rebounded and related factors during positive airway pressure therapy. Scientific Reports, 11, Article 7686. https://doi.org/10.1038/s41598-021-87149-3
16. Sládek, M., Klusáček, J., Hamplová, D., & Sumová, A. (2023). Population-representative study reveals cardiovascular and metabolic disease biomarkers associated with misaligned sleep schedules. Sleep, 46(4), zsad037. https://doi.org/10.1093/sleep/zsad037
17. Vallat, R., Shah, V. D., & Walker, M. P. (2023). Coordinated human sleeping brainwaves map peripheral body glucose homeostasis. Cell Reports Medicine, 4(7), Article 101100. https://doi.org/10.1016/j.xcrm.2023.101100
18. Windred, D. P., Burns, A. C., Lane, J. M., Saxena, R., Rutter, M. K., Cain, S. W., & Phillips, A. J. K. (2024). Sleep regularity is a stronger predictor of mortality risk than sleep duration: A prospective cohort study. Sleep, 47(1), zsad253. https://doi.org/10.1093/sleep/zsad253
19. Yan, P., Feng, S., Ma, M., Li, B., & Liu, J. (2026). Summary of the best evidence that cognitive behavioral therapy for insomnia improves sleep quality in patients with chronic insomnia. Frontiers in Psychiatry, 16, Article 1688561. https://doi.org/10.3389/fpsyt.2025.1688561
20. Yoshikawa, M., Yamauchi, M., Fujita, Y., Ono, M., Ushijima, S., Kinoshita, T., & Kimura, H. (2020). Dynamic changes in nocturnal blood glucose levels are associated with sleep-related features in patients with obstructive sleep apnea. Scientific Reports, 10, Article 17873. https://doi.org/10.1038/s41598-020-74908-x
21. Zhu, S., Shi, X., Chen, M., Shi, Y., Li, Y., Zheng, X., & Li, Y. (2025). The effect of sleep disruption on cardiometabolic health. Life, 15(1), Article 60. https://doi.org/10.3390/life15010060
22. Winkelman, J. W., Berkowski, J. A., DelRosso, L. M., Koo, B. B., Scharf, M. T., Sharon, D., Zak, R. S., Kazmi, U., Falck-Ytter, Y., Shelgikar, A. V., Trotti, L. M., & Walters, A. S. (2025). Treatment of restless legs syndrome and periodic limb movement disorder: An American Academy of Sleep Medicine clinical practice guideline. Journal of Clinical Sleep Medicine, 21(1), 137–152. https://doi.org/10.5664/jcsm.11390
23. Maki, P. M., Panay, N., & Simon, J. A. (2024). Sleep disturbance associated with the menopause. Menopause, 31(8), 724–733. https://doi.org/10.1097/GME.0000000000002386
24. Sparks, J. R., Burgess, H. J., & colleagues. (2025). Menopause-related changes in sleep and the associations with cardiometabolic health: A narrative review. Healthcare, 13(17), 2085.
25. Gardiner, J., et al. (2025). Systematic review and meta-analysis examining alcohol and sleep in healthy adults.
26. McCullar, J., et al. (2024). Presleep alcohol consumption and objective sleep architecture outcomes. Sleep, 47(4).
27. Deantoni, M., Reyt, M., Dourte, M. et al. Circadian rapid eye movement sleep expression is associated with brain microstructural integrity in older adults. Commun Biol 7, 758 (2024). https://doi.org/10.1038/s42003-024-06415-y
28. Feriante J, Singh S. REM Rebound Effect. [Updated 2024 Sep 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560713/?utm_source=chatgpt.com
29. Greenhill, C. Feasibility and effectiveness of time-restricted eating in different populations. Nat Rev Endocrinol 18, 715 (2022). https://doi.org/10.1038/s41574-022-00770-8
Just finished reading all the three sleep articles.
What a fantastic comprehensive summary on sleep you have written...starting with Sleep scores, interpreting the wearable sleep data, decoding various sleep stages and then a 'Masterclass' on sleep disruption levers and a superb yet simple framework to approach those disruptions with a practical approach.
This is truly a 'Masterclass' in sleep that every physician should read. The way you integrated Circadian biology into Sleep as an output was just a scientific marvel. I really liked the way you described 'Peripheral clocks' which cannot perceive Light as the 'Central SCN clock' does, but uses food cues to entrain with the Brain clock ( Brain-Organ clock cross-talk) I would encourage every physician no matter their speciality to read your write-up on sleep series. Thank you for writing such a marvelous series on this well needed education on Sleep. Real Kudos to you !
Thank you for doing this article. Very clear and clinically useful.
Out of curiosity, how does sleep architecture change with normal ageing, and how might that interact with the three drivers you describe?