

How the body tells time
A mechanistic tour of circadian timekeeping for clinicians and the curious-minded


Part one of two. This collaborative piece stays inside the mechanism: how a cluster of neurons and a loop of genes generate, distribute, and fine-tune biological time. A warning in advance, this can get pretty technical but because education should be accessible for all, there are blocked sections along the way that summarize the technical bits so do not despair. There is a little something here for everyone.
Part two will be taking this machinery into the clinic.
Sleep is the most visible output of the circadian system, but it is only one expression of a much larger physiological timing network. Circadian rhythms are intrinsic, near-24-hour oscillations that organize physiology at the molecular, cellular, and organ-system levels. Their purpose is not to coordinate sleep. It is to organize physiology in time [1,2].
The feature worth highlighting is that this system is fundamentally anticipatory rather than reactive.
We are trained to think of physiology as a sequence of responses:
Glucose rises, and insulin is released.
A pathogen appears, and immune cells respond.
Tissue is damaged, and repair pathways activate.
Circadian biology runs on a different logic. Many of its processes begin preparing for predictable demands before those demands arrive [2,5]. In, the circadian world:
Insulin secretory capacity increases ahead of habitual meal times.
Immune surveillance follows daily rhythms tuned to the active phase.
DNA repair and other maintenance programs ramp up during rest, when energy can be diverted from immediate survival toward housekeeping.
Cortisol begins climbing in the hours before waking, priming the transition from sleep to activity.
Anticipation is the whole game. The body is betting on the day ahead, but it can’t place that bet unless it knows what time it is. And it turns out a pile of cells has no obvious way of knowing that.
So how does it tell time?
The body needs a reference clock
At the center of the system sits a cluster of roughly 20,000 neurons in the anterior hypothalamus: the suprachiasmatic nucleus (SCN) [6].
The SCN is routinely called the body’s ‘master clock,’ which is useful but slightly misleading because it is not the only clock. It is the central coordinator for a vast network of peripheral clocks distributed across nearly every tissue.
The liver keeps time. So do the pancreas, skeletal muscle, adipose tissue, and immune cells [1]. Even the gut microbiome shows circadian rhythmicity that feeds back into our physiology.
The SCN, of course, does not perform any of the downstream work but what it does contribute is phase: a shared rhythmic reference the body can check in with. Like an orchestra conductor, it plays no instrument; it sets the beat that the section players keep.
The metaphor invites an obvious question: what makes the conductor trustworthy?
Why is the SCN a more reliable clock than, say, the liver? Not because its neurons are better oscillators. Individually, they’re rather poor at oscillation. They are noisy, imprecise, and prone to drifting apart and fading out within a few cycles if left alone in a petri dish.
The SCN’s reliability emerges from the cells’ ability to continuously signal to one another and pull each other back onto a common phase. That mutual entrainment (coupling) is what converts a population of mediocre individual clocks into one robust, self-correcting rhythm.
Our anatomy makes this possible. The core of the SCN is where the light signal lands, delivered directly from the retina by the retino-hypothalamic tract (RHT), while the shell sits downstream and relays the timing onward. Light enters at the core, and corrected phase information propagates out from there.
The cohesion that binds the whole network is largely a single dominant signal: VIP (vasoactive intestinal peptide), released by core neurons and received across a cell’s nucleus through its receptor. VIP signaling is what keeps thousands of single-cell oscillators locked to a shared phase.
Remove VIP and the SCN fractures, the neurons scatter into incoherent individual rhythms, and the body loses its consolidated daily pattern of activity and rest [6].
A whole host of additional signals help to fine-tune that cohesion:
GRP (gastrin-releasing peptide) provides a redundant photic channel
GABA (γ-aminobutyric acid) adjusts phase moment to moment
AVP (arginine vasopressin) in the shell lends the network its rigidity, which is part of why jet lag is so stubborn [6].
But the headline is this: the cross-talk yokes thousands of imperfect oscillators into one coherent, self-sustaining rhythm that no single neuron could produce alone. The SCN’s role as pacemaker is a property of the entire network, not of any cell within it.
That robustness is exactly what other tissues, lacking this dense coupling, do not have. It’s why the liver needs a conductor.
Hold onto the conductor analogy, because it’s the key to this article: most circadian pathology is not a broken instrument. It’s a roomful of musicians playing the right tune at the wrong time.
How cells build a 24-hour day
How does any individual cell tell time at all? Although the SCN does act as the central clock, the beat itself doesn’t originate there. Let’s clarify.
The capacity to oscillate doesn't originate in the SCN at all. Essentially, every nucleated cell can keep time on its own. The same molecular clock ticks in a liver cell as in an SCN neuron. What the SCN uniquely provides isn't the oscillation; it's the coordination.
Thousands of cells can each run a clock, but only a densely coupled population can hold them all to the same time and broadcast it to the rest of the body. The SCN is the central clock, not because it's the only one, but because it's the one the others set their proverbial watch by.
That time-telling mechanism is the transcription–translation feedback loop (TTFL). At its core are several clock genes:
CLOCK (Circadian Locomotor Output Cycles Kaput)
BMAL1 (Brain and Muscle ARNT-Like 1)
Period (PER1/2/3), and
Cryptochrome (CRY1/2) [3,4]
Bear with me, this is where we’re getting technical. I’ve added a higher-level overview just afterward to explain in accessible terms.
CLOCK and BMAL1 are transcription factors that partner and bind E-box motifs in the promoters of the PER and CRY genes, driving transcription.
PER and CRY proteins accumulate in the cytoplasm, are progressively phosphorylated, and translocate back to the nucleus as a bulky PER:CRY:CK1 repressor complex. This is where CRY makes direct contact with CLOCK: BMAL1 to inhibit it while PER scaffolds the assembly, shutting down the very dimer that produced them.
Negative feedback in its purest form.
As the repressors are phosphorylated and degraded, the brake lifts, CLOCK: BMAL1 re-engage their E-boxes, and the cycle restarts [4].
That core loop doesn’t run alone, and the second loop is quite magical. CLOCK: BMAL1 also transcribe two opposing families of nuclear receptors (REV-ERBα/β and RORα/β/γ), which then compete at ROR response elements in the BMAL1 promoter, REV-ERBs repressing and RORs activating.
This drives BMAL1’s own rhythm in near-antiphase to PER/CRY and is a major reason the oscillation is stable rather than fragile [3,4] (REV-ERB is heme-responsive and druggable, which is why it surfaces constantly in the metabolic-clock and pharmacology literature).
A further output tier (the DBP/E4BP4 pair acting on D-box elements) relays clock timing onto thousands of downstream genes that carry no E-box of their own [4].
In other words: Picture a tiny machine inside every nucleated cell in your body. Two proteins team up and switch on a handful of genes, and those genes build a second pair of proteins whose only job is to switch the first pair back off.
So the ‘machine’ spends all day building its own off-switch. Once enough of the ‘off’ proteins pile up, they shut the whole thing down. Then the cell slowly clears them away, the switch flips back on, and the cycle starts over.
That build-up-and-clear-out loop takes almost exactly twenty-four hours, and that, ticking away in cell after cell, is the clock. A second, slower loop runs alongside it like a counterweight, keeping the rhythm steady instead of wobbly.
A visual walkthrough of the process is below as Figure 1. Feel free to skip right past it if this level of detail makes your eyes glaze over; nothing later depends on it. If you'd like it as a separate, high-resolution download to keep or print, just leave a comment at the bottom.
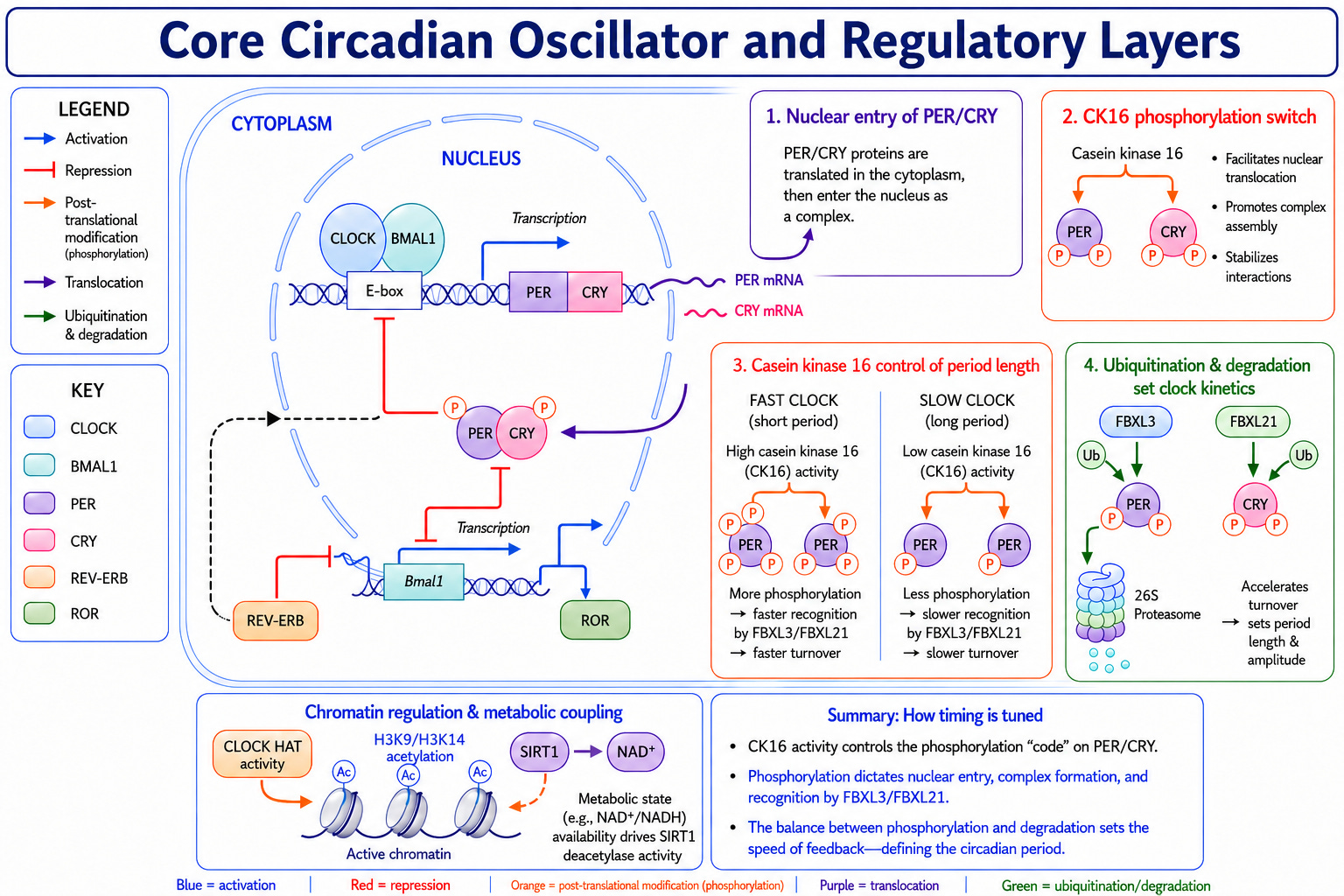
The molecular detail is quite elegant, but the key concept that matters is much simpler: The clock is a biochemical delay circuit.
If repression immediately followed transcription, the system would collapse into equilibrium. Oscillation requires lag, and lag is exactly what the chemistry supplies because the entire process of transcription, translation, protein folding, phosphorylation, nuclear import, and regulated degradation each takes time.
The ~24-hour period is the emergent sum of those cumulative delays [4].
It is a genuinely different way to think about physiology. The clock isn’t reading time off some master standard tucked away in the hypothalamus.
There is no standard.
The interval is the chemistry: built, fresh, every single day, out of how long it takes proteins to be made, folded, tagged, shipped, and destroyed. The clock does not measure time. It creates it.
Why the clock resets every day
Here is the fact that makes the rest of the system necessary, and what the ‘master clock’ language papers over: the human circadian oscillator has a free-running period that averages close to 24.2 hours [2,6].
Park a person in constant darkness, and their clock drifts about twelve minutes later every day. It doesn’t sound like much, but run this experiment for a week, and the person would slide into a different time zone without leaving the room. Run it long enough, and they will be sleeping through the afternoon.
Turns out, the clock by itself runs quite slowly, and that slowness compounds.
So the clock must be corrected daily against an external signal. Any environmental cue capable of resetting it is called a zeitgeber (German for ‘time-giver’), and the dominant zeitgeber for the SCN is light.
This daily correction is entrainment, and its direction depends entirely on when the light arrives. Light in the morning produces a phase advance, shifting the clock earlier and pulling physiology forward to meet the day. Light in the evening produces a phase delay, shifting the clock later.
The same light stimulus can impose opposite effects, governed by the biological time (each individual’s internal clock) of the day. Plotted across the cycle, this relationship is the phase response curve (PRC):
Light in the late night and early morning causes advance,
Light in the evening and early night causes a delay, and
Light through the middle of the subjective days is neutral territory [2,6].
The PRC is arguably the single most actionable mechanistic concept in the field, because it explains why light is not simply “good” or “bad.” A bright morning is therapeutic for a delayed clock but counterproductive for an advanced one.
How light becomes biology
Let’s trace the light input pathway at the molecular level, because it’s where light literally becomes transcription. Intrinsically photosensitive retinal ganglion cells (ipRGCs) express melanopsin, an opsin maximally sensitive to short-wavelength light near 480 nm. Morning light.
Where rod and cone opsins mediate image-forming vision, melanopsin signals through a separate cascade that directly depolarizes the ipRGCs. These cells are able to detect light (photoreceptive), which is why circadian photoreception can survive in forms of blindness that abolish vision [2]. Their axons bundle into the RHT and run straight to the SCN core as a single, direct connection from eye to clock.
What happens at that synapse is the resetting event, and it’s surprisingly concrete.
The terminals of the RHT release two transmitters: glutamate, which carries the ‘light is happening’ signal, and PACAP (pituitary adenylate cyclase-activating polypeptide), which modulates the gain.
Together, they tune how large a shift a given light pulse produces.
Glutamate opens NMDA (N-methyl-D-aspartate) receptors, and this is the pivotal conversion: the receptor admits a slug of Ca²⁺, turning a synaptic signal into an intracellular one.
Calcium is therefore the message.
That calcium then activates two kinase relays, whose job is to carry the signal from the membrane to the nucleus and amplify it along the way [the kinases are both mouthfuls: CaMKII (calcium/calmodulin-dependent protein kinase II and the MAPK/ERK cascade (mitogen-activated protein kinase / extracellular signal-regulated) kinase].
Applying this to the PRC, then, light in the early night drives clock proteins up while the loop is winding down, delaying the clock; the same pulse in the late night drives it up as the loop is already ramping, advancing it and pushing sleep time later.
When evening light reaches the SCN, it suppresses pineal melatonin output and nudges the clock later, in effect insisting that dusk hasn’t arrived yet [2,5]. For nearly all of human history, that was honest information where sunrise and sunset were the signal.
The same circuitry now stares into screens, LEDs, and buildings that never go dark. The SCN never got the memo that any of this is artificial. To it, a photon is a photon, and a phone at midnight is just a very small, very confusing sunrise.
So, how sensitive is this system? Early research found that ordinary room light (around 100 lux) was enough to cut melatonin roughly in half [7].
More recent work pushed that number even lower for the average person: light dimmer than a bedside lamp, about the brightness of a phone screen, around 30 lux [8].
But the real surprise wasn't the average; it was how wildly people differ. Some individuals' melatonin drops by half under barely any light at all, while others can tolerate fifty times more before seeing the same effect [8].
In practice, that means the same late-night scrolling that wrecks one person's sleep signal barely registers for another. There's no single "safe" level of evening light that holds for everyone
This is also why melatonin, beyond its traditional sleep role, is the most useful phase marker we have. Measured under dim light, the dim-light melatonin onset (DLMO) is the current gold standard for clocking an individual’s internal phase, and the reason circadian research is so fanatical about keeping study lighting below ~30 lux.
Timing is more than transcription
The transcription-translation feedback loop, happening with the core clock genes, sets up the basic oscillation for peripheral tissue. There is a second regulatory layer that determines how precise and how fast that oscillation runs.
Making the clock proteins is only half the story.
Once PER and CRY exist, what really sets the clock's speed is how they're chemically modified after the fact, and especially how quickly they're tagged for destruction [4].
The key enzymes are kinases. They phosphorylate PER at two competing sites: one marks it for rapid breakdown by the cell's protein-disposal system, shortening its lifespan, and the other stabilizes it to slow the breakdown. The balance between those two reactions acts like a molecular dimmer switch on the clock's pace.
It also explains a rather elegant trick. The clock keeps near-perfect time across a range of body temperatures, because the two opposing reactions speed up and slow down in tandem and cancel out [4,5].
CRY is regulated the same way, by a separate set of proteins that compete to either destroy it or shield it; tilting that balance lengthens or shortens the day directly [4].
The recurring theme is noteworthy: the clock's tempo is set less by how fast its genes are switched on than by how fast its braking proteins are cleared away.
The clinical proof of concept is familial advanced sleep phase syndrome (FASPS): affected individuals fall asleep in the early evening and wake spontaneously hours before dawn.
The first family identified with this syndrome carried a single change in one of the PER proteins, swapping one amino acid for another at exactly the stabilizing site described above, so the protein is broken down too quickly [5].
A second family had a mutation in the kinase itself, arriving at the same result from the other direction. In both, the clock was fully built and structurally normal. The only thing that changed was how fast one protein was processed, and that was enough to shift the entire daily schedule hours earlier.
The machine is working perfectly for these patients, but the tempo is off.
FASPS is the legible version of something that happens to all of us, all the time. Phosphorylation state, protein stability, and degradation rate are nudged continuously by metabolic status, aging, environmental exposure, and (increasingly the interesting part) drugs.
The takeaway for the clinic is almost philosophical: the clock is not a fixed structure you either have or lack. It’s a tempo, constantly being set, and big changes in someone’s sleep and behavior can fall out of changes in molecular kinetics.
A clock inside the genome
Just when you thought we were done with regulatory mechanisms, there is yet another layer. Seems there’s always another system with the system, and this one sits right on the border between circadian biology and epigenetics.
As it turns out, the clock doesn’t just run in the cell. It controls the door to the genome.
For a gene to be transcribed, its surrounding chromatin must be accessible. DNA wound tightly around histones is effectively hidden; DNA in an open configuration is available. Given everything above, it should be no surprise that these accessibility states oscillate across the day.
Here, the clock does something more clever than just switching genes on and off. The same machinery that activates clock genes also loosens the way DNA is packaged around them, and DNA has to be unpacked before it can be read. So as the clock runs, it's continually opening and closing access to different stretches of the genome, on a daily schedule.
Genes that are readable in the morning may be tucked away by night.
When you look across all the organs in the body, roughly 40% of our genes run on a daily rhythm somewhere, so this isn’t a niche effect. Determining which gene cycles depends entirely on the tissue [12]. The clock is essentially putting a daily timestamp on close to half the genome [3].
This cracks open one of the most satisfying puzzles in the field.
The liver and the immune system run the exact same core clock. Same genes, same loop. Yet they do completely unrelated jobs. How?
The clock isn’t the difference. The genome it’s knocking on is.
Each tissue brings its own chromatin architecture, its own transcription factors, its own enhancer repertoire, so the identical timing signal reaches an entirely different set of genes.
The clock simply tells it when, and the tissue decides what. Circadian rhythms, then, are conducting whole transcriptional programs across the genome, tissue by tissue (which is why the orchestra metaphor is so hard to retire.)
Every tissue keeps its own time
Now we have determined that peripheral tissues contain their own autonomous molecular clocks, built on the same TTFL architecture but wired into tissue-specific transcription factors, chromatin landscapes, and metabolic signaling [1].
Liver, pancreas, skeletal muscle, adipose, gut, and immune cells all run from the same blueprint and produce completely different outputs:
In the liver, clock oscillations interlock with regulators of gluconeogenesis, glycogen handling, lipid metabolism, bile acid synthesis, and xenobiotic detoxification.
In pancreatic β-cells, clock-controlled genes time insulin secretion and glucose responsiveness.
In skeletal muscle, circadian signaling integrates with energy-sensing pathways (notably AMPK) and mitochondrial bioenergetics to shape fuel selection and performance capacity.
In the immune system, clocks intersect with cytokine networks, glucocorticoid signaling, and immune-cell trafficking to set inflammatory tone across the day [1,2].
How does the master clock keep all these scattered tissue clocks on the same schedule? It uses the signals the whole body already shares.
Cortisol is a major one: the brain's clock drives its daily rise and fall, and that rhythm reaches almost every tissue, nudging each local clock into line. The daily swing in core body temperature does similar work, as do the autonomic nervous system, the timing of meals, and melatonin at night [1,2].
The liver is the interesting exception because it listens less to cortisol and more to when you eat, which is exactly why meal timing turns out to matter so much (more on that in part two). Together, these body-wide cues keep every local clock locked to the same time of day, even as each tissue runs its own distinct program.
The functional payoff is temporal sequencing. Digestion, nutrient handling, immune surveillance, cellular repair, and cardiovascular load are not run simultaneously; they are spread across the day according to demand.
The body is therefore organized both anatomically and rhythmically.
The clock and metabolism are in constant dialogue
The relationship between the clock and metabolism runs in both directions: the clock regulates metabolism, and metabolism regulates the clock.
So far, the clock has looked like it runs the show. But the traffic goes both ways, and the body's energy state talks back to the clock just as forcefully. The cell tracks its fuel status partly through NAD⁺, and that signal is wired directly into the clock machinery, adjusting it according to whether energy is abundant or scarce.
The clock also controls how NAD⁺ is made, so each governs the other (yes, yet another feedback loop, but by now that's the point: this system is loops within loops, top to bottom).
Exercise is the clearest example. Muscle contraction burns through fuel, and the energy-sensing pathways that detect this reach in and change how quickly core clock proteins are broken down. This is a direct line from how hard a muscle is working to the timing of its clock.
Eating, fasting, exercise, and overall energy availability are all inputs and signals that physically reshape the clock's own parts.
This stops being abstract in human muscle, where the metabolism–clock loop has been demonstrated directly. Collecting biopsies several times across the day and under controlled conditions, van Moorsel and colleagues showed that human muscle exhibits a genuine day–night rhythm in mitochondrial oxidative capacity, peaking in the late afternoon to early evening.
And this happens independent of recent activity or feeding [9]. The mitochondrion, in other words, has a schedule.
That rhythm is not fixed for life. It is blunted with insulin resistance, where disrupted muscle clock oscillations track with altered rhythmic mitochondrial metabolism [9], and it is dampened in older, metabolically compromised individuals [11], illustrated in Figure 2, below:
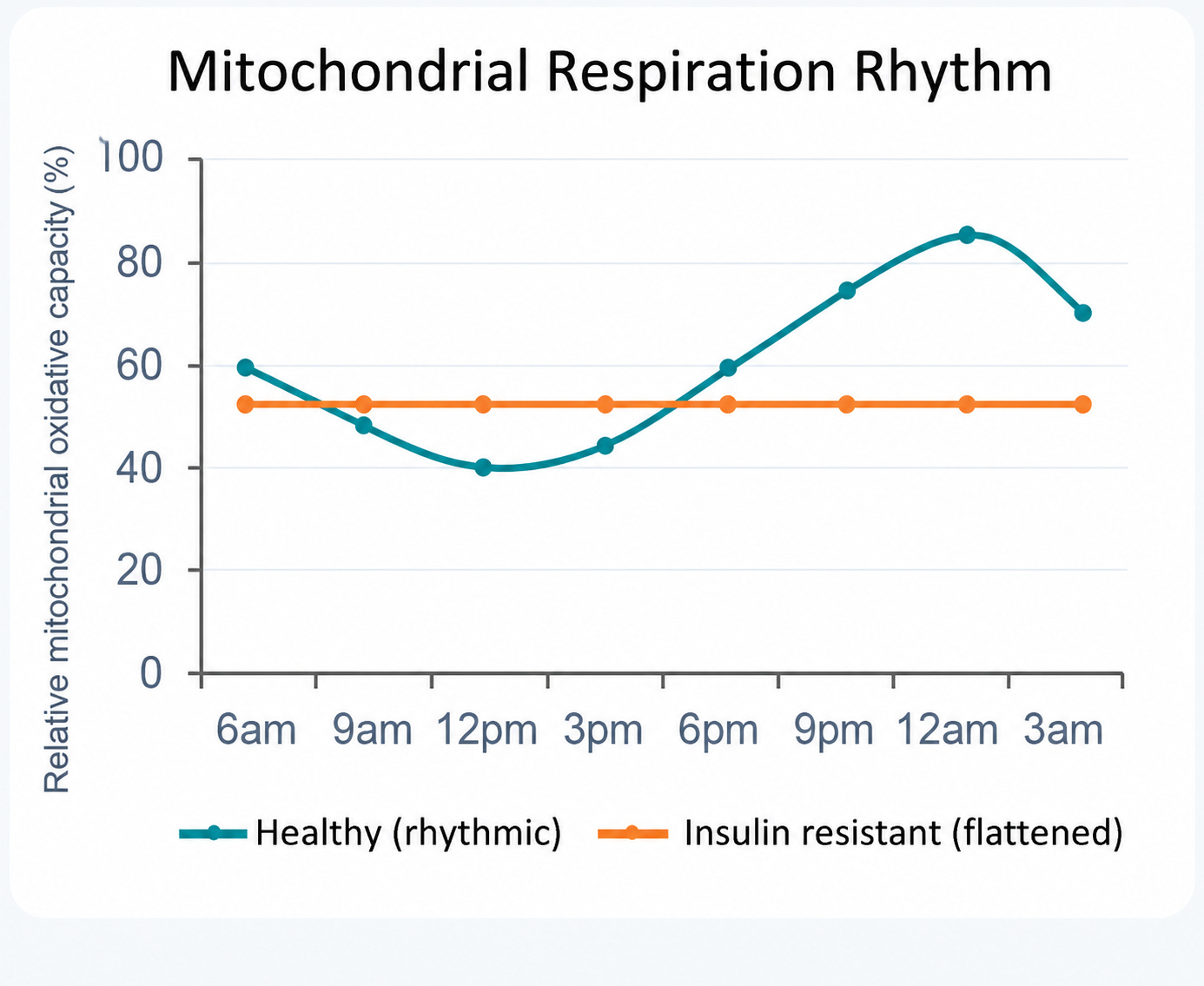
Whether the loss of rhythm is cause, consequence, or both remains an open and clinically loaded question, but mechanistically the lesson is consistent with everything above: when the timing degrades, the machinery may look intact while the output flattens.
What this buys us
Strip it all the way down, and one principle is left standing. The circadian clock is a self-sustaining biochemical oscillator corrected daily by light, copied into every tissue, gated through chromatin, tuned by kinases, and in constant back-and-forth with metabolism. Out of that architecture, the body pulls off something physiology otherwise can’t: it acts before it’s acted upon. It shows up early to the party.
The pattern keeps repeating, and when ‘dysfunction’ is present, it’s almost never that the clock is broken. The parts work. The timing slipped.
That’s not a pedantic distinction; it’s the hinge the entire next article swings on. Because if the failure mode is timing rather than hardware, then timing is something we can actually get our hands on.
That’s where part two goes. We will be getting out of the molecular weeds and into the clinic: how the inputs impact the orchestra, meal timing, light hygiene that respects the phase response curve, melatonin’s life beyond sleep, shift work and social jet lag, chronotype, and the internal desynchrony that may link all of them to chronic disease.
References
Bautista, J., Ojeda-Mosquera, S., Ordóñez-Lozada, D., & López-Cortés, A. (2025). Peripheral clocks and systemic zeitgeber interactions: From molecular mechanisms to circadian precision medicine. Frontiers in Endocrinology, 16, Article 1606242. https://doi.org/10.3389/fendo.2025.1606242
Campbell-Galland, A., Bafna, A., & Jagannath, A. (2025). The molecular circadian clock: From fundamental mechanisms to therapeutic promise in neurological disorders. Advanced Drug Delivery Reviews, 224, Article 115653. https://doi.org/10.1016/j.addr.2025.115653
Fagiani, F., Di Marino, D., Romagnoli, A., et al. (2022). Molecular regulations of circadian rhythm and implications for physiology and diseases. Signal Transduction and Targeted Therapy, 7(1), 41. https://doi.org/10.1038/s41392-022-00899-y
Laothamatas, I., Rasmussen, E. S., Green, C. B., & Takahashi, J. S. (2023). Metabolic and chemical architecture of the mammalian circadian clock. Cell Chemical Biology, 30(9), 1033–1052. https://doi.org/10.1016/j.chembiol.2023.08.014
Liu, Y., Huo, R., & Zhang, E. E. (2025). Evolving perspectives on the molecular and neural foundations of mammalian circadian rhythms. Trends in Neurosciences, 48(11), 904–918. https://doi.org/10.1016/j.tins.2025.09.009
Patton, A. P., & Hastings, M. H. (2023). The mammalian circadian time-keeping system. Journal of Huntington’s Disease, 12(2), 91–104. https://doi.org/10.3233/JHD-230571
Zeitzer, J. M., Dijk, D. J., Kronauer, R. E., Brown, E. N., & Czeisler, C. A. (2000). Sensitivity of the human circadian pacemaker to nocturnal light: melatonin phase resetting and suppression. The Journal of Physiology, 526(Pt 3), 695–702. https://doi.org/10.1111/j.1469-7793.2000.00695.x
Phillips, A. J. K., Vidafar, P., Burns, A. C., et al. (2019). High sensitivity and interindividual variability in the response of the human circadian system to evening light. Proceedings of the National Academy of Sciences, 116(24), 12019–12024. https://doi.org/10.1073/pnas.1901824116
van Moorsel, D., Hansen, J., Havekes, B., Scheer, F. A. J. L., Jörgensen, J. A., Hoeks, J., Schrauwen-Hinderling, V. B., Duez, H., Lefebvre, P., Schaper, N. C., & Schrauwen, P. (2020). Human skeletal muscle exhibits a day–night rhythm in mitochondrial oxidative capacity. Proceedings of the National Academy of Sciences, 117(15), 8758–8764. https://doi.org/10.1073/pnas.1916823117
Gabriel, B. M., Altintas, A., Smith, J. A. B., et al. (2021). Disrupted circadian oscillations in type 2 diabetes are linked to altered rhythmic mitochondrial metabolism in skeletal muscle. Science Advances, 7(43), eabi9654. https://doi.org/10.1126/sciadv.abi9654
Wefers, J., Connell, N. J., Fealy, C. E., et al. (2020). Day-night rhythm of skeletal muscle metabolism is disturbed in older, metabolically compromised individuals. Molecular Metabolism, 41, 101050. https://doi.org/10.1016/j.molmet.2020.101050
Zhang, R., Lahens, N. F., Ballance, H. I., Hughes, M. E., & Hogenesch, J. B. (2014). A circadian gene expression atlas in mammals: Implications for biology and medicine. Proceedings of the National Academy of Sciences, 111(45), 16219–16224. https://doi.org/10.1073/pnas.1408886111
 | A guest post by
|